
Spinoserebellaariset ataksiat eli SCA-sairaudet
- CD–10: G11.8 Muut määritetyt perinnölliset ataksiat, G11.9 Määrittämätön perinnöllinen ataksia, ORPHA:99
Spinoserebellaariset ataksiat eli SCA-sairaudet ovat useimmiten aikuisiällä alkavia eteneviä hermoston rappeutumiseen johtavia sairauksia. SCA-sairauden oireet johtuvat ensisijaisesti pikkuaivojen (-cerebellum) ja selkäytimen (spina-) hermojen toimintahäiriöistä. SCA-sairaudet periytyvät aina autosomisesti vallitsevasti eli dominantisti (AD-periytyminen).
Ataksialla tarkoitetaan tahdonalaisten liikkeiden hallinnan ja koordinoinnin ongelmia. Ataksiasairauksien taustalla on toimintahäiriö pikkuaivoissa tai niiden hermoratayhteyksissä (Kuva 1.). Pikkuaivot sijaitsevat aivojen takaosassa. Pikkuaivoissa on kaksi rakenteellista osa: pikkuaivopuoliskot (hemisfäärit) ja niitä yhdistävä pikkuaivomato (vermis). Hemisfäärien kuorikerros (pikkuaivokuori) muodostuu hermosoluista ja ydinosa hermojen viejähaarakkeita (aksoneita) suojaavasta valkeasta aineesta (myeliinistä).
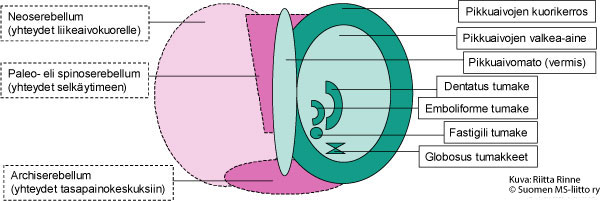
Kuva 1. Pikkuaivojen rakenne. Kuvassa oikealla on pikkuaivojen kehitysopilliset alueet ja vasemmalla pikkuaivojen liikesäätelyyn osallistuvat rakenteet.
Pikkuaivojen rakenne
Kehitysopillisesti pikkuaivojen vanhin alue (Archiserebellum) on yhteydessä sisäkorvien tasapainoelimeen ja silmänliikkeiden säätelykeskuskuksiin. Kehitysopillisesti toiseksi vanhin pikkuaivorakenne on paleoserebellum. Paleoserebellum sijaitsee pikkuaivohemisfääreissä molemmin puolin pikkuaivomatoa (vermis). Paleoserebellum vastaanottaa liike- ja asentoviestejä selkäytimestä, josta syystä aluetta kutsutaan spinoserebellumiksi. Häiriöt spinoserebellumissa heijastuvat seisomatasapainossa, kävelyssä (kävelyataksia) ja vartalonhallinnassa (vartaloataksia).
Pikkuaivojen hemisfäärit (Neoserebellum) ovat vahvasti poimuttuneita ja edustavat hermoston kehityksessä nuorinta vaihetta. Neoserebellum on vain nisäkkäillä. Molemmilla pikkuaivokuorilla on tarkasti rajoittuneet alueet, jotka ovat yhteydessä isojen aivojen vastaaviin liikekeskuksiin (motoriset kuorialueet). Nämä yhteydet välittyvät ydinjatkeessa sijaitsevien alempien oliva-tumakkeiden kautta (Nucleus olivae inferior). Oliva-tumakkeista lähtevien kiipeävien hermosyiden (climbing fibers) aksonit risteävät ennen saapumistaan pikkuaivokuorelle. Kiipeävät hermosyyt säätelevät pikkuaivojen kuorikerroksen Purkinjen solujen aktiivisuutta. Niiden toiminta aiheuttaa äkillisen, lyhytkestoisen aktivaation Purkinjen soluissa. Purkinjen soluista lähtevät viejähaarakkeet päätyvät pikkuaivotumakkeisiin (Pikkuaivojen nukleukset), joita on neljä molemmissa pikkuaivokupoleissa. Fastigii-tumakkeen toiminta osallistuu kehon ja raajojen molemminpuolisten lihasliikkeiden säätelyyn. Globosus- ja emboliforme-tumakkeet säätelevät kehon samanpuoleisia lihasliikkeitä. Dentatus-tumakkeet koordinoivat samanpuoleisia hienomotorisia liikkeitä. Pikkuaivotumakkeiden viejähaarakkeet ovat myös yhteydessä väliaivojen talamus-tumakkeisiin (Nucleus ventralis lateralis). Talamus-tumakkeesta lähtee hermoyhteyksiä isoaivokuorien liikekeskuksiin sekä Ruber-tumakkeisiin. Ruber-tumakeesta saa alkunsa selkäytimen rubrospinaalinen hermorata. Talamus-tumakkeen kautta pikkuaivojen toiminta puolestaan synkronisoituu toisen liikesäätelyn kannalta merkittävän hermojärjestelmän – tyvitumakkeiden (basaaligangliot) kanssa.
Pikkuaivojen toiminta
Pikkuaivojen keskeiset tehtävät liittyvät tasapainon ylläpitämiseen ja liikkeiden koordinointiin. Ne osallistuvat tahdonalaisten liikkeiden täsmälliseen ajoitukseen, rytmitykseen kuin myös liikekaarien laajuuksien hallintaan. Pikkuaivovauriossa tasapaino heikkenee ja käsien osumatarkkuus heikkenee – ne haparoivat kohteen ohi ja ympäri, tapailevat kohdettaan (ataksia). Ataksia todetaan selvimmin nopeissa liikkeissä. Liikettä suorittavien (agonistien) ja niitä vastustavien lihasten (antagonistien) yhteistoiminta hidastuu. Tästä seurauksena on dysdiadokokinesia, mikä havaitaan esim. käden nopeassa pyöritysliikkeessä. Liikkeiden pysäyttäminen ei onnistu (rebound-ilmiö): antagonististen lihasten toiminta ei riitä hidastamaan agonistien toimintaa. Pikkuaivot huolehtivat monimutkaisten liikesarjojen suorittamisesta, ”muistissa pysymisestä” eli automaattisista liikesuorituksista ja ne osallistuvat uusien motoristen taitojen omaksumiseen. Äkillisesti alkaneessa pikkuaivovauriossa toimintahäiriöt todetaan samalla puolella raajoissa.
Etenevä pikkuaivosairaus
Etenevissä pikkuaivosairauksissa ensioireina ovat ongelmat nopeiden, tarkkojen, yhtäaikaisten liikkeiden suorittamisessa, tasapainon ylläpitämisessä ja uusien liiketaitojen oppimisessa. Liikesuoritukset, jotka edellyttävät kaikkien neljän raajan yhtaikaista toimimista tai molempien käsien saumatonta työskentelyä eivät onnistu. Pikkuaivosairauden ensivaiheessa tasapainovaikeuksien ohella todetaan hienomotoriikan ongelmia ja ongelmia jo opittujen liiketaitojen suorittamisessa (esim. juokseminen, pyörällä ajaminen, tanssiminen jne.). Uusien liiketaitojen oppiminen on työlästä, eikä sairauden edetessä ole enää mahdollista.
Silmien liikehäiriöt sekä silmänliikkeiden seuranta- ja kohdistusvaikeudet ovat usein pikkuaivosairauksiin liittyviä oireita samoin puheen kangertaminen ja äänen voimakkuuden vaihtelu (dysartria). Pikkuaivoista on myös hermoyhteyksiä niin oppimista kuin käyttäytymistä sääteleviin aivojen otsalohkoihin (frontoserebellaariset radat).
SCA-sairauksissa todetaan joko yksinomaan etenevä pikkuaivovauriosta johtuva oireisto (tasapaino- ja liikkeiden koordinointivaikeus, silmien liikehäiriöt ja artikulaation ongelmat) tai lisäksi laaja-alainen etenevä niin isojen aivojen, selkäytimen kuin ääreishermojenkin toimintaa vaurioittava neurologinen oireisto (Ataksia + muut neurologiset oireet). Sairastavan oireiden ja neurologisen tutkimuksen perusteella saadaan tietoa sairauden aiheuttamista vaurioista ja niiden laajuudesta hermostossa (Kuva 2):
a) sairaus vaurioittaa vain pikkuaivojen toimintoja (serebellaarinen ataksia)
b) sairaus vaurioittaa sekä pikkuaivojen että ääreishermoston toimintoja (serebellaarinen ataksia + tunto- ja ääreisliikehermojen vaurio eli polyneuropatia)
c) pikkuaivojen ja ylemmän liikeradan vaurio (ataksia + pyramidiratavaurio)
d) sairaus vaurioittaa kaikkia kohdissa 1–3 mainittuja hermoyhteyksiä sekä aiheuttaa muutoksia mahdollisesti myös käytöksessä, toimintojen suunnittelussa, muistissa
e) sairaus vaurioittaa kaikkia kohdissa 1–4 mainittuja hermoyhteyksiä sekä aistinelimiä (erityisesti näköä tai kuuloa).
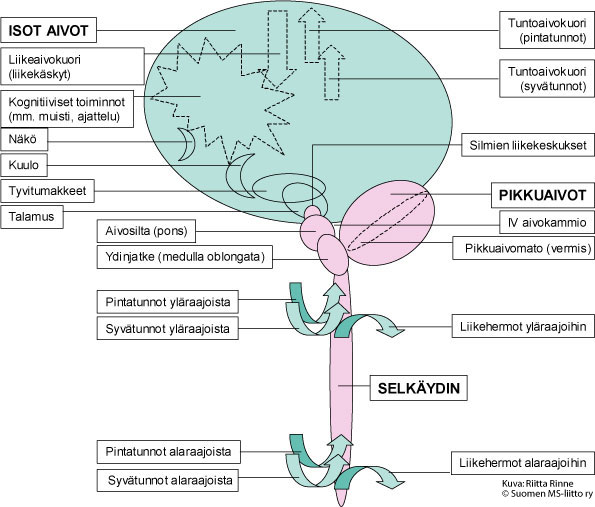
Kuva 2. Hermoston rakenne. Pikkuaivot ovat yhteydessä hermoston muihin rakenteisiin.
SCA-sairaudet ja niiden diagnoosi
Ensimmäisen autosomisesti vallitsevasti eli dominantisti periytyvän ataksiasairauden (ADCA-sairaus) kuvasi tohtori Menzel vuonna 1893. Perheenjäsenillä todettiin vaihtelevasti löydöksiä niin pikkuaivojen, aivorungon kuin selkäytimen vaurioista. Suvuittain esiintyviä ja autosomisesti dominantisti periytyviä SCA-sairauksia on sittemmin kuvattu lukuisia eri puolilla maailmaa ja niiden ryhmittely on ollut ja on edelleen kirjavaa. Neuropatologisia muutoksia (hermosolujen rappeutumaa eli atrofiaa) SCA-sairauksissa on todettu ensisijaisesti pikkuaivojen ja aivorungon alueella sekä selkäytimessä. Neuropatologisten löydösten perusteella sairauksia on aikaisemmin kutsuttu OPCA-sairauksiksi (Olivopontocerebellaarinen atrofia).
1980-luvun alkupuolella englantilainen neurologi Anita Harding loi käyttökelpoisen kliinisen ryhmittelyn vallitsevasti periytyville ataksiasairauksille: ADCA tyypit I – VI (Taulu 1). Tämä ryhmittely on auttanut SCA-sairauksien geenimuutosten selvitystyössä, joka mahdollistui 1990-luvulla. SCA-sairaudet ovat osoittautuneet geneettisesti varsin moninaiseksi sairausryhmäksi.
SCA-sairauden kliininen diagnoosi perustuu neurologiseen tutkimukseen. Sitä täydentävät muiden erikoisalojen konsultaatiot (silmälääkäri tai neuro-oftalmologi, korvalääkäri tai neuro-otologi, neurofysiologi, neuroradiologi). Perinnöllisyyslääkärin neuvonta sairastavalle ja hänen sukulaisilleen on aina aiheellista.
Aivojen kuvantamistutkimuksissa todetaan vaihtelevasti hermosolujen rappeutumista (atrofiaa) pikkuaivoissa, aivorungossa ja selkäytimessä. Ääreishermojen tutkimuksessa (ENMG-tutkimus) ja hermojen keskushermostossa tapahtuvaa viestintää eli herätepotenteeleja (SSEP-tutkimus) voidaan todeta muutoksia. SCA-sairauksissa tavanomaiset verikokeet ja selkäydinnestetutkimukset ovat normaaleja. Osaan SCA-sairauksista on käytettävissä sairauden mutaation tunnistava DNA-testi, joka varmistaa diagnoosin. SCA-sairaus voidaan joskus DNA-tutkimuksella todeta potilaalta, jonka suvussa SCA-sairautta sairastavia ei ole diagnosoitu. Sairautta kutsutaan tällöin sporadiseksi. Sporadinen geenivirhe periytyy seuraavaan sukupolveen AD-periytyvän sairauden periytymisen lainalaisuuksia noudattaen.
SCA-sairauksia aiheuttavat useat eri geenimutaatiot
Ensimmäinen SCA-sairautta aiheuttava geenimuutos tunnistettiin kromosomissa 6 vuonna 1993. Toistaiseksi sairauksia aiheuttava geenivirhe tai geenivirheen paikannus on tehty jo lähes kolmessakymmenessä SCA-sairaudessa (SCA1, SCA2, SCA3 …SCA28 ja DRPLA). Tauluun 1 on sijoitettu nyt tunnistetut SCA-sairaudet ja sairauden ADCA-tyypin I – VI mukainen oirekuva. Muistettava on, että samaa SCA-sairautta sairastavien perheenjäsenten oireissa ja niiden etenemisessä esiintyy usein huomattavaa vaihtelua.
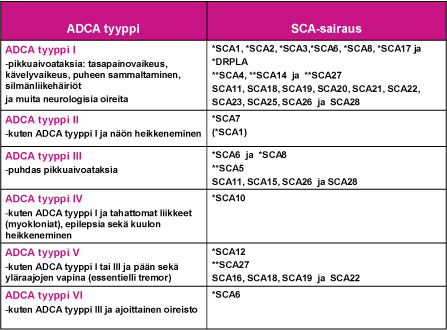
Taulu 1. SCA-sairauksien ryhmittäminen ADCA-tyyppeihin I – VI.
Selitykset:
- SCA = sairauden aiheuttavan geenin paikannus on tehty, mutta geenimutaatio on toistaiseksi tunnistamatta
- *SCA = sairautta aiheuttava geenimutaatio tunnetaan ja se on määritettävissä suoraan verinäytteestä DNA-tutkimuksella, joka on käytettävissä sairauden kliinisessä erotusdiagnostiikassa.
- **SCA = sairautta aiheuttava geenimutaatio tai saman geenin useampia sairautta aiheuttavia mutaatioita tunnetaan, DNA-tutkimus ei toistaiseksi ole käytettävissä tavanomaisessa kliinisessä erotusdiagnostiikassa.
SCA-sairauden geenimutaation tunnistaminen on edellytys geenin ohjaaman valkuaisaineen määrittämiselle. Viidessä ensimmäiseksi tunnistetussa SCA-sairauden geenissä (SCA1, SCA2, SCA3, SCA6 ja DRPLA) mutaatio johtui kolmen nukleotidiemäksen (CAG) CAG-jakson pidentymisestä. CAG-jakso ohjaa valkuaisaineessa glutamiini nimisen aminohapon tuotantoa. Myös terveillä SCA-potilaan sukulaisilla oli kyseisissä geeneissä CAG-jakson monistuma. Sairauden ilmaantumisen kannalta keskeiseksi CAG-jakson pituus: sairastuneilla CAG-jaksojen määrä oli merkittävästi kasvanut. Pidentynyt CAG-jakso ohjaa ylipitkän glutamiiniketjun synnyn valkuaisaineeseen. Niitä SCA-sairauksia, joiden geenivirheenä todetaan ylipitkä CAG-jakso, kutsutaan polyglutamiiniataksioiksi (PolyQ-ataksiat). Sittemmin myös SCA7- ja SCA17-sairaudessa on todettu pidentynyt CAG-jakso. Pidentynyt CAG-jakso ei ole ainoa SCA-sairautta aiheuttava geenimutaatio.
SCA8-, SCA10- ja SCA12-sairauden mutaatioissa on niin ikään todettu pidentynyt nukleotidiemäsjakso: SCA8:ssa CTG-jakso, SCA10:ssa ATTCT-jakso ja SCA12:ssa CAG-jakso. Näissä sairauksissa mutaatio sijaitsee geenin luentakehyksen ulkopuolella. Seurauksena ei ole geenin ohjaaman valkuaisaineen kertyminen soluihin, vaan pikemminkin ongelma kyseisen valkuaisaineen tuottamisessa. Muissa tähän mennessä tunnistetuissa SCA-mutaatioissa ei ole todettu nukleotidijaksojen pidentymistä. Sairauksien (SCA4, SCA5, SCA13, SCA14 ja SCA27) tunnistetut mutaatiot johtavat virheellisen, toimimattoman tai puutteellisesti toimivan valkuaisaineen syntyyn.
Taulukko 2. Kootusti tietoa SCA-sairauksien kromosomipaikannuksista, geenimutaatioista sekä geenituotteesta eli valkuaisaineesta (proteiinista).
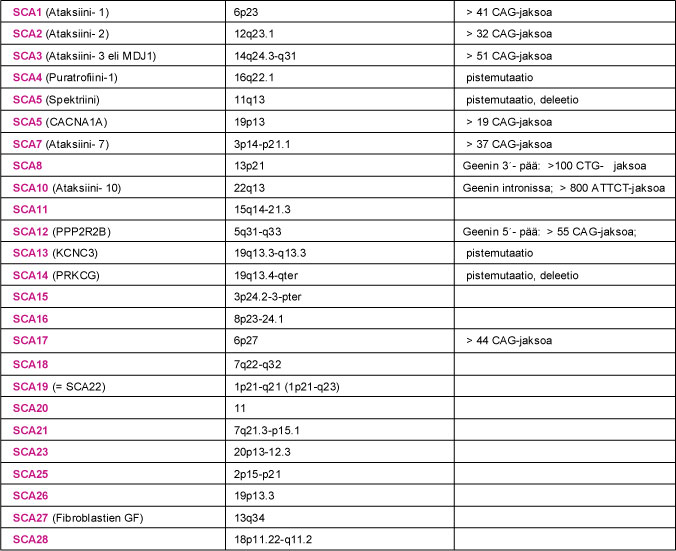
Taulu 2. SCA-sairaus, valkuaisaine (proteiini), sairautta aiheuttavan geenin kromosomialue ja tunnetut geenimutaatiot. SCA9- ja SCA24-sairauksista ei ole toistaiseksi tietoja käytettävissä.
Erilaiset hermosolujen toimintahäiriöt aiheuttavat ataksiasairauden
Perustutkimuksen antama tieto SCA-sairauksien ja muiden ataksiaa aiheuttavien sairauksien solutason häiriöistä on johtamassa uuteen ataksiasairauksien ryhmittämiseen. A Fillan työryhmän (2004) ehdottamassa uudessa ryhmittelyssä lähtökohtana on solun toimintahäiriö. Uuden ryhmittelyn tarkoituksena on selventää ataksiasairauksien toisistaan poikkeavia sairausmekanismeja sekä luoda ymmärrystä ja pohjaa ataksiasairauksien lääkehoitojen kehittämiseen.
Nyt tunnetut SCA-sairaudet sijoittuisivat uudessa ryhmittelyssä kolmen eri pääryhmän alle.
Alla ataksiasairauksien pääryhmät (Ryhmät 1–6) ja SCA-sairaudet. A Fillan työryhmän ehdotus ataksiasairauksien uudesta ryhmittelystä.
Mukaeltu: De Michele G et. Al., J Neurol 251: 913-922, 2004.
- RYHMÄ 1: Mitokondriaaliset ataksiat
- RYHMÄ 2: Aineenvaihduntahäiriöistä johtuvat ataksiat
- RYHMÄ 3: Puutteellisesta DNA:n korjaantumisesta johtuvat ataksiat
- RYHMÄ 4: Proteiinikertymistä ja proteiinin hajottamisongelmista johtuvat ataksiat
- Polyglutamiiniataksiat (SCA1, SCA2, SCA3, SCA6, SCA7, SCA17 ja DRPLA)
- RYHMÄ 5: Ionikanavasairauden aiheuttamat ataksiat
- Kalium-kanava: EA1 ja SCA13
- Kalsium-kanava: EA2 ja EA5
- RYHMÄ 6: Muut perinnölliset ataksiat
- AD-ataksiat, joissa mutaationa on toistojaksopidentymä geenin luentakehyksen ulkopuolella (SCA8, SCA10, SCA12) tai muu tunnettu mutaatio (SCA4, SCA5, SCA14 ja SCA27)
- AD-ataksiat, joiden geenialue on paikannettu, mutta mutaatio tunnistamatta (SCA11, SCA15, SCA16, SCA18, SCA19, SCA20, SCA21, SCA22, SCA23, SCA25, SCA26 ja SCA28)
SCA-sairauksien esiintyvyys
Suomessa yleisimmiksi ovat osoittautuneet SCA8 ja SCA7. SCA1 on 3. tavallisin, sen sijaan SCA2, SCA6 ja SCA17 diagnoosit ovat erittäin harvinaisia. SCA-sairauksia arvellaan maailmanlaajuisesti olevan 1–5 / 100 000. Nyt käytettävissä olevin diagnoosimenetelmin varmistunee SCA-sairautta aiheuttavan geenin varmistuminen noin 60–70 %:lla. Yleisin SCA-sairaus on SCA3, joka edustaa noin 20–50 % kaikista SCA-sairauksista eri puolella maapalloa. Brasiliassa SCA3 on todettu jopa 85 %:lla SCA:n syyksi. Suomessa ei toistaiseksi SCA3:a ole diagnosoitu. Seuraavaksi yleisimmät SCA-sairaudet ovat SCA2 (13 – 18%) ja SCA1 (10%). Erittäin harvinaisia SCA-sairauksia ovat SCA11, 13, 15, 16, 18, 19, 20, 21, 22, 23, 25, 26 ja 28. Näissä sairauksissa sairastavien oiretiedot perustuvat yhteen tai kahteen kuvattuun sukuun.
SCA-sairauksien oireet muistuttavat toisiaan
SCA-sairauksien kliininen erotusdiagnoosi on ongelmallista. Silmänliikehäiriöiden laatu (SCA1, SCA2, SCA3 ja SCA6), ääreishermostovaurion (SCA2, SCA3, SCA4,SCA8, SCA12, SCA18, SCA25, SCA27), pyramidirata- (SCA1, SCA3, SCA7) tai ekstrapyramidirataoireiden (SCA2, SCA17, DRPLA) toteaminen voivat auttaa kliinisessä epäilyssä ja ”täsmägeenitestin” valinnassa. SCA-sairaudet alkavat pääsääntöisesti keski-ikäisenä, mutta SCA1:n, SCA2:n, SCA3:n, SCA7:n, SCA13:n, SCA17:n ja DRPLA:n oireet voivat alkaa jo lapsuusvuosina. SCA6 ilmenee usein vasta myöhäisellä keski-iällä (> 45 vuoden iässä). SCA6- ja SCA8-diagnooseja on todettu myös potilailla, joiden sukulaisilla ei ole diagnosoitua ataksiasairautta eli sporadisina.
SCA-sairauksien hoidosta
SCA-sairauksiin ei ole toistaiseksi lääkehoitoja. Lääkityksellä voidaan vaikuttaa joihinkin SCA-sairaudessa esiintyviin oireisiin (mm. levottomat jalat, rakon toimintahäiriöt, epilepsia, tahattomat liikkeet). Sairastunutta ja hänen läheisiään voidaan auttaa ja tukea huolehtimalla tarvittavan niin ammatillisen, lääkinnällisen kuin sosiaalisen kuntoutuksen suunnittelemisesta sekä kuntoutuksen asianmukaisesta ja oikea-aikaisesta järjestämisestä.
Kirjoittaja
Riitta Rinne, LL, neurologian erikoislääkäri
Maskun neurologinen kuntoutuskeskus
Julkaistu 21.06.2006
Kirjallisuusviitteet
Kirjallisuusviitteet SCA-katsaus_2006
Oppaat
Ataksiaoireyhtymät. Tietoa etenevistä ataksiasairauksista. Riitta Rinne ja Eila Niemi. Suomen MS-liitto, 1996.
Järjestötoiminta
Potilasjärjestö: Neuroliitto ry; ataksiaverkosto
Muita tietolähteitä
The National Ataxia Foundation: www.ataxia.org
Sveriges socialstyrelsens kunskapsdatabas om ovanliga diagnoser innehåller information om fler än 300 ovanliga sjukdomar och tillstånd.